United States: Food and Drug Administration (FDA)
The Digital Health Center of Excellence (DHCoE) is part of the planned evolution of the Digital Health Program in the Center for Devices and Radiological Health (CDRH) and will align and coordinate digital health work across the FDA. It marks the beginning of a comprehensive approach to digital health technology, setting the stage for advancing and realizing the potential of digital health.
The DHCoE provides regulatory advice and support to the FDA’s regulatory review of digital health technology and is not responsible for making marketing authorization decisions. Key documents include:
United Kingdom: National Institute for Health and Care Excellence (NICE)
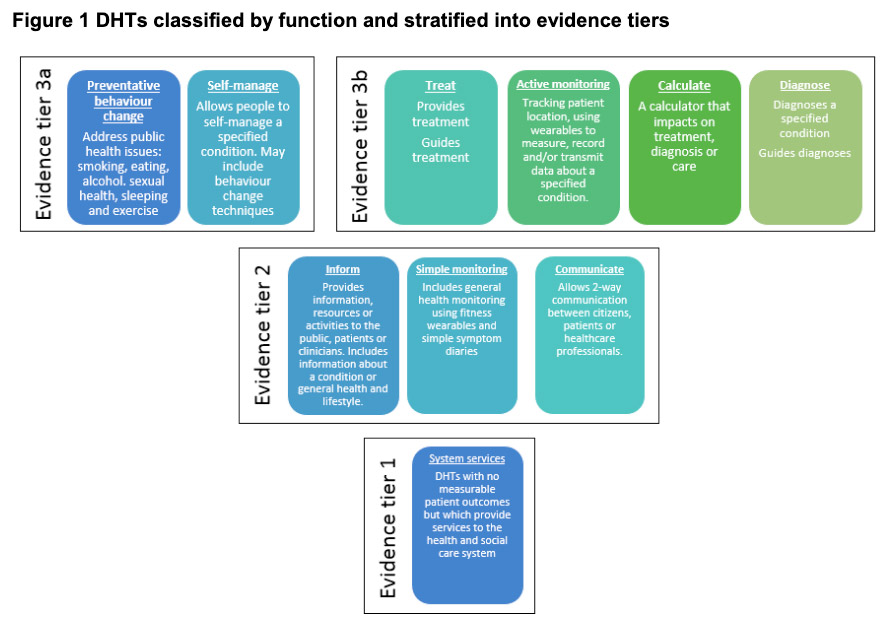
NICE provides national guidance and advice to improve health and social care. As digital health technologies develop at an increasing pace, they worked with partners to develop standards that ensure new technologies are clinically effective and offer economic value.
The aim of NICE’s Evidence for Effectiveness standards is to make it easier for innovators and commissioners to understand what good levels of evidence for digital healthcare technologies look like. Digital healthcare technologies must also meet the needs of the health and care system, patients, and users.

(Source)
Germany: Federal Institute for Drugs and Medical Devices
The Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte, BfArM) is an independent federal higher authority within the portfolio of the German Federal Ministry of Health.
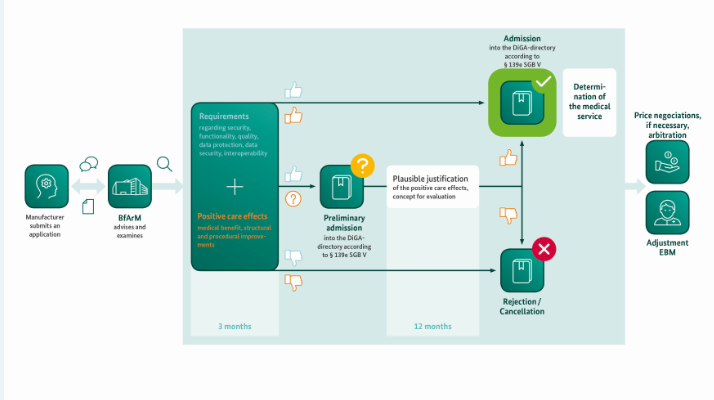
BfArM’s Assessment Procedure is designed as a fast-track process: Within a three-month period at the most, starting with the filing of the complete application, BfArM will the DiGA, a subset of which include DTx products. The essence of this assessment is the examination of the manufacturer’s statements about the product qualities – from data protection to interoperability and user friendliness – and the examination of the evidence of the positive healthcare effect of the DiGA provided by the manufacturer. These are effects through which the state of a patient’s health or his/her possibilities for dealing with his/her disease are improved by the use of the DiGA. (Source)
Additional resources include: